Blog
Long-Term Retention of Bioanalytical Watson LIMS™ Studies: A Compliance-First Perspective
Most bioanalytical labs running Watson LIMS™ have never made a conscious decision about archiving. They have simply deferred. What 21 CFR Part 11 and EU GMP Annex 11 actually require, and why format independence is a regulatory posture, not an IT preference.

Executive Summary
The majority of bioanalytical laboratories running Thermo Watson LIMS™ have, by default, adopted a data retention strategy that was never intended as a long-term archive. Keeping Watson™ running because completed studies live inside it is not an archive strategy. It is a liability that compounds every year.
This paper examines three questions that QA Directors and Bioanalytical Lab Directors working with Watson™ environments regularly face:
- What do 21 CFR Part 11, EU GMP Annex 11, and ICH M10 actually require of a long-term study archive?
- Does Watson™’s built-in archiving functionality satisfy those requirements?
- What does a regulator-ready, vendor-independent long-term archive look like, and how is data integrity maintained across a 15-year retention horizon?
The regulations do not require completed studies to remain in the system that generated them. They require records to be accurate, complete, human-readable, and verifiable over time. A format-independent archive with cryptographic integrity verification fully satisfies that obligation. Whether compliance is robust or merely contingent depends entirely on the format chosen.a compliance decision, not an IT decommissioning decision. And the format in which data is archived determines whether that compliance is robust or contingent.
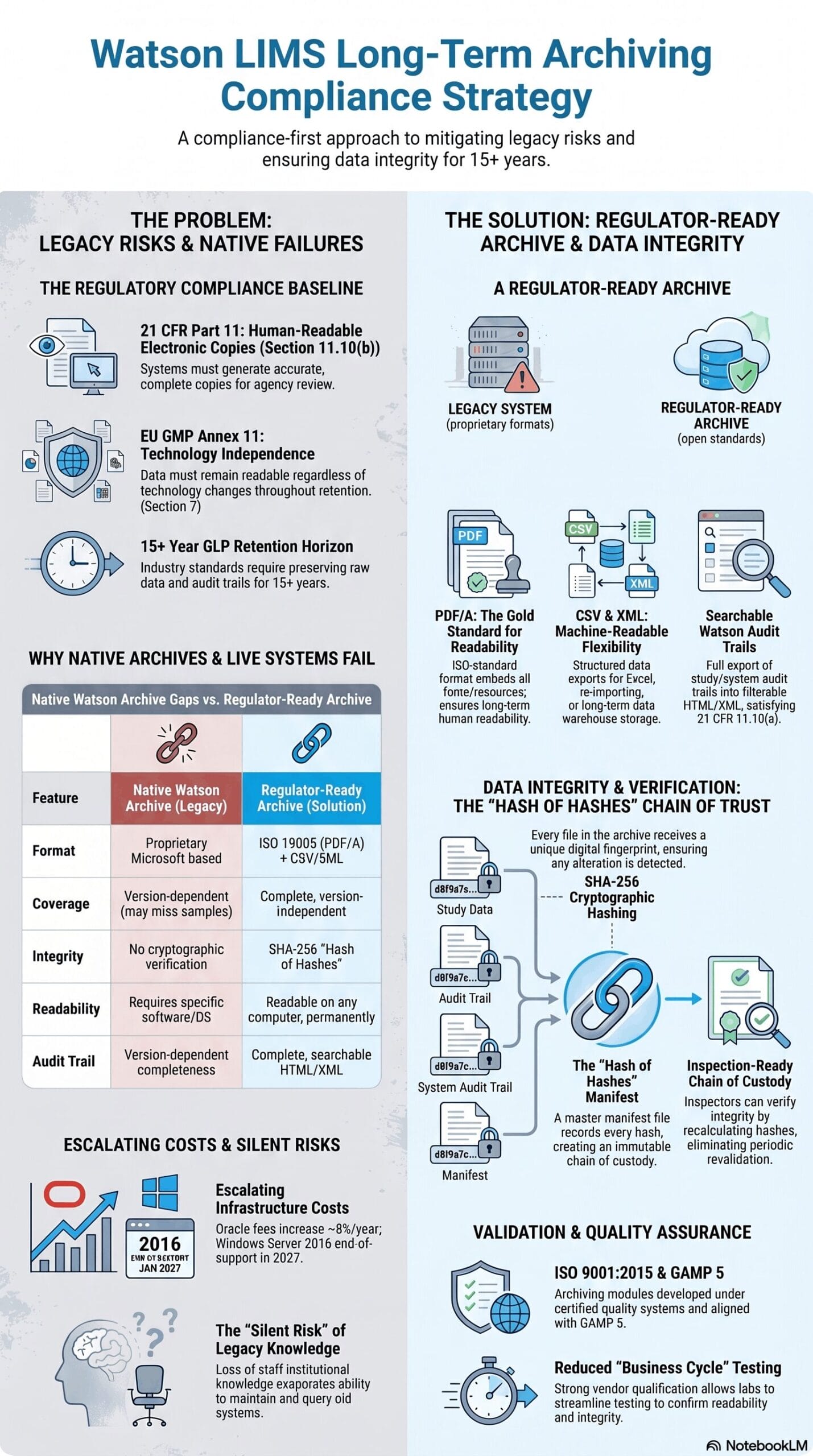
The Compliance Baseline: What the Regulations Actually Require
Before evaluating any archiving approach, it is worth reading the relevant regulatory text directly, rather than through the lens of received interpretations.
Data Integrity and Long-Term Archiving: Aligning ICH M10 with 21 CFR Part 11.10(b)
ICH M10 requires full documentation of all validation data and analytical results, including failed or out-of-specification runs. This is reinforced by 21 CFR Part § 11.10(b), which mandates that regulated systems generate accurate and complete copies of records in both human-readable and electronic formats suitable for agency inspection.
Crucially, 21 CFR Part § 11.10(b) defines the output requirement rather than the specific hosting system. A structured archive of PDF/A, CSV, and XML files satisfies these standards independently of Watson™, Oracle™, or any proprietary viewer. This distinction is operationally vital: while maintaining Watson™ is one path to compliance, a system-agnostic archive is often a more robust strategy for ensuring data integrity over a decades-long retention horizon.
Regulatory Alignment
21 CFR Part 11 § 11.10(e) and EU GMP Annex 11 § 7 require complete, unfiltered audit trails that remain accessible and readable over time. This includes capturing all Watson™ events, such as study reopens, result rejections, and reassays, without summarizing the data.
An archive fulfills this requirement by capturing the full Watson™ audit trail, including user identity, timestamps, and actions such as study reopens, result rejections, and reassays. Compliance depends on completeness: the archive must preserve exactly what Watson™ held, without filtering, aggregation, or summarization.
Long-Term System Independence
While 21 CFR § 58.195 sets a baseline GLP retention period of 2 to 5 years, industry practice extends to 15 years or longer. Over this horizon, data must remain readable independent of specific versions of Watson™, Oracle™, or Windows Server, making technical format independence the central engineering challenge.
“But Watson™ Already Has a Study Archive”: Why That Is Not Enough
This is the first response most QA teams give when the subject of archiving is raised, and it deserves a direct answer. Watson™ does include a Study Archive function. For teams that have not closely examined their technical characteristics, it is easy to assume that running Watson Study Archive™ satisfies the long-term retention requirement. The gaps are real and material.
Structural Vulnerabilities of Proprietary Exports
Many quality assurance teams assume the built-in Watson Study Archive™ meets long-term regulatory requirements. However, technical checks show major data gaps.
Export completeness depends entirely on the software version used. In several versions, the tool completely excludes critical Sample Information. This includes sample identity, matrix, nominal concentration, and related metadata.
Without this data, you cannot answer regulatory questions about sample handling, reassays, or exclusions. This creates a major compliance risk once that specific software version is no longer supported by the vendor.
The Compliance Risks of Software Dependency
The Watson Study Archive™ export utilizes a proprietary Microsoft format rather than an ISO-standardized preservation format, providing no guarantee of long-term readability. Conversely, ISO 19005 (PDF/A) ensures multi-decade legibility by embedding all required rendering resources directly within the file, a standard this proprietary format fails to meet.
This dependency violates EU GMP Annex 11 § 7, which requires that records must remain readable regardless of technology changes. For a 15-year retention period, the native tool introduces a critical compliance risk by making data survival contingent on software stability outside your control.
The Silent Risk: Completed Studies Still Living in Production Systems
The most common long-term retention approach in bioanalytical laboratories is to leave completed datasets in the active Watson™ production environment, allowing technical debt, licensing overhead, and operational vulnerabilities to accumulate undetected.
Infrastructure Dependency
Watson™ explicitly relies on a Microsoft Windows environment for its application layer and a relational Oracle Database™ backend. Maintaining this architecture introduces compounding support costs, as Oracle™ licensing fees escalate by approximately 8% annually. Furthermore, underlying enterprise operating systems such as Windows Server 2016 reach the end of mainstream support in January 2027, causing Extended Security Update costs to double annually thereafter. Combined with standard 5-to-7-year GxP hardware refresh cycles, labs that defer decommissioning face compounded infrastructure obsolescence and security vulnerabilities.
Knowledge Loss and Corporate Mergers
Maintaining legacy application environments depends on a small group of specialists who understand proprietary schema configurations and system validation histories. Staff turnover drastically inflates the operational overhead of querying the database during regulatory inspections, whereas migrating to a vendor-neutral repository embeds this metadata directly into the files. This system independence is critical during pharmaceutical M&A, where GxP regulations prohibit decommissioning duplicate Watson™ environments without a compliant archive in place, stalling software consolidation windows.
The Inspection Scenario
Regulatory authorities can issue data queries regarding bioanalytical studies more than a decade after completion, mandating the prompt generation of compliant, inspectable records. Querying a legacy system that has not been formally maintained, patched, or validated introduces severe technical and data-integrity risks. This operational compliance risk is completely eliminated by migrating the assets into system-agnostic, open-standard file formats at study completion.
What a Regulator-Ready Archive Looks Like: Format and Content
To satisfy 21 CFR Part 11 §§ 11.10(b), (e) and EU GMP Annex 11 § 7, an archive must simultaneously deliver absolute completeness, format independence, and verifiable integrity through open, system-agnostic formats:
- PDF/A (ISO 19005): Eliminates external dependencies by embedding all fonts and color profiles, ensuring files render identically for decades without proprietary viewers. Files are organized by analytical run and by study to streamline inspection workflows.
- CSV Data Streams: Every dataset (including analytical results, calibration curves, regressions, QC data, failed runs, rejected samples, reassays, and deactivation comments) is exported to CSV. Data represents the final Watson™ status without filtering or summarization to ensure absolute fidelity.
- XML Architecture: Deliver self-describing, plain-text datasets optimized for programmatic access, multi-study cross-analysis, and seamless re-importing into downstream platforms like a new LIMS, eTMF, or data warehouse.
- Audit Trail Exports: The complete study-level and system-level Watson™ audit trail is exported in HTML/XML format, capturing every user identity, timestamp, and action type without aggregation, thereby directly satisfying 21 CFR § 11.10(e).
- SHA-256 Cryptographic Verification: Integrity is verified by generating a unique SHA-256 hash for each exported file. These hashes are compiled into a master hash-of-hashes manifest, ensuring any post-archive modification is instantly detectable.
Archival Scope Boundaries
While an automated archive handles the core data export, the regulated company plays an essential role in managing specific record types that live outside the standard Watson™ study file. Addressing these elements ensures complete compliance and a seamless decommissioning process:
- Chromatography Data: Raw instrument files from data acquisition systems (such as Empower, MassHunter, or Analyst) live in their own repositories rather than inside Watson™, which only stores the integrated final numbers. The company must ensure that these raw source files are preserved in accordance with standard laboratory data retention policies.
- System-Level Data: Global items, such as reference standards and system-wide assay batch records, sit outside individual study boundaries. The company can easily run a targeted workflow to capture these system-level logs before turning off the older legacy servers.
Archive Format Comparison
| Feature | Watson Study Archive™ (built-in) | Regulator-Ready Archive (PDF/A + CSV + XML) |
|---|---|---|
| Data Coverage | Version-dependent; omits Sample Information in multiple releases | Complete, version-independent export keeping Sample Information intact |
| Format Longevity | Proprietary Microsoft format requiring active legacy software | ISO 19005 (PDF/A) and open CSV/XML, readable on any computer, permanently |
| Integrity Control | Lacks built-in cryptographic verification | Secured via per-file SHA-256 hashes and a master manifest |
| Audit Trail | Completeness depends on the active software version | Complete, unfiltered HTML/XML audit trail export |
Cryptographic Data Integrity: Answering the Inspector’s Hardest Question
A structured archive addresses format longevity but does not automatically prove that files remain unaltered over time. To address regulatory queries about data tampering, the archiving architecture establishes a secure chain of custody using SHA-256 hashing.
The SHA-256 Verification Process: How It Works
During archive creation, the system generates a unique SHA-256 digital fingerprint for every individual file. These fingerprints are compiled into a master archive manifest. The manifest itself is then hashed to create a secure “hash of hashes,” which is recorded in two independent locations: within the archive package and inside the system processing protocol log.
To verify archive integrity at any point during the 15-year retention period, a user recalculates the file hashes and compares them to the master manifest. A complete match proves that no data has been modified, added, or deleted.
Audit Readiness and Infrastructure Migration
Inspectors can verify archive integrity on demand via hash comparison, replacing assertions of trust with reproducible technical proof. Because a static archive cannot alter itself between reads, periodic revalidation is eliminated, and storage migrations require only a single verification step.
In Practice: Boehringer Ingelheim
Boehringer Ingelheim exemplifies a compliance posture that is increasingly common among large pharmaceutical organizations that have systematically addressed the long-term retention question.
Rather than waiting for a decommissioning trigger or a regulatory pressure event, Boehringer archives completed Watson™ studies upon completion using the StudyPlus Archive Module, developed by up to data GmbH. A specialist in regulatory study data management for pharmaceutical and life sciences companies, up to data GmbH has supported the automation of laboratory processes for over 20 years. The archive exists independently of the Watson™ environment from the moment it is created, satisfying the retention obligation regardless of what subsequently happens to Watson™, Oracle™, or the underlying Windows infrastructure.
This is structurally different from keeping Watson™ running and treating continued system availability as the mechanism for satisfying retention. One approach creates a permanent dependency on a live system. The other eliminates it. The compliance posture may look identical on paper. The risk profile is not.
Peer reference available. Organizations evaluating their archiving options can speak directly with Boehringer Ingelheim through the up to data GmbH reference program.
Validation Considerations
For QA teams evaluating any new process or system, the validation question arises early. In the case of a static-output archiving module, the answer is more straightforward than most.
Vendor Qualification Under GAMP 5
The StudyPlus Archive Module is vendor-qualified to support a GAMP 5 validation process, and up to data GmbH is ISO 9001:2015 certified. The module has been tested and validated against Watson™ 7.3, 7.4, and 7.6.
Under GAMP 5 qualification, the customer’s internal validation obligation can be reduced to a business-cycle test: confirming that a representative set of known studies can be located in the archive, that their content is complete and readable, and that hash verification passes. This is substantially less than a full IQ/OQ/PQ cycle, and the supplier’s qualification documentation is available as part of the engagement.
No Periodic Revalidation for a Static Archive
A validated running system requires periodic revalidation because its configuration can change, its software is updated, and its processes can drift. A hash-verified static archive has none of these characteristics. There is no configuration to maintain, no update cycle, and no process that could alter the archive content between reads.
The hash manifest verifies integrity at the point of creation; that verification can be re-run at any time as part of an inspection preparation, a storage migration, or a scheduled data integrity review. It does not constitute revalidation. It is a verification operation.
This is one of the less obvious advantages of a static-file archive over a retained live system: the validation burden does not compound over time. A running Watson™ environment requires ongoing validation and maintenance. An archive does not.
Decommissioning Documentation
Retiring a validated GxP system requires documented evidence that the process was executed correctly and that data integrity was maintained throughout. A complete package consists of a Decommissioning Specification, Execution Records, and a Decommissioning Report. The StudyPlus Archive Module generates a processing protocol for each archive run in StudyReporter, its integrated reporting component, and this protocol forms part of the execution record. Documentation templates structured to meet these requirements are available as part of the engagement.
Conclusion: The Archive Is a Compliance Decision, Not a Default
Most organizations have never consciously decided how to retain completed Watson™ studies. They have simply left them in the system that generated them. That is a compliance posture. It is just one that was never made deliberately, and whose costs and risks rarely surface until they become acute.
21 CFR Part 11 and EU GMP Annex 11 do not require completed studies to remain in the system that generated them. They require records to be accurate, complete, and human-readable across the defined retention period, with demonstrable integrity. A well-constructed archive of ISO-standardized open formats with cryptographic verification fully satisfies those requirements. Keeping Watson™ alive does not satisfy them in a unique way.
The Watson Study Archive™ is a useful operational tool. It is not a substitute for a format-independent, integrity-verified archive. Its coverage is version-dependent, its output format is proprietary, and its long-term readability depends on technology continuity that no vendor can guarantee across a 15-year horizon.
The practical question is not whether to archive. It is when. Organizations that archive at study completion carry the lowest risk at the lowest cost. The archive exists from day one. The retention obligation is met from day one. Whatever decisions follow about Watson™, Oracle™, Windows, or the broader infrastructure are unconstrained by compliance risk.
The format chosen for archiving determines whether compliance is robust or contingent on systems, vendors, and software versions that will not outlast the retention obligation.
References
European Commission (2011). EU guidelines for good manufacturing practice for medicinal products for human and veterinary use: Annex 11, computerized systems. URL: https://health.ec.europa.eu/system/files/2016-11/annex11_01-2011_en_0.pdf.
European Medicines Agency & Pharmaceutical Inspection Co-operation Scheme (2025). Joint stakeholders consultation on the revision of Chapter 4 on Documentation, Annex 11 on Computerized Systems, and on the new Annex 22 on Artificial Intelligence of the PIC/S and EU GMP Guides. PIC/S. URL: https://picscheme.org/en/news/joint-stakeholders-consultation-on-the-revision-of-chapter-4.
International Organization for Standardization (2005). ISO 19005-1:2005: Document management — Electronic document file format for long-term preservation — Part 1: Use of PDF (PDF/A). URL: https://www.iso.org/standard/38920.html.
International Society for Pharmaceutical Engineering (2022). GAMP 5: A risk-based approach to compliant GxP computerised systems (2nd ed.). URL: https://ispe.org/publications/guidance-documents/gamp-5-guide-2nd-edition.
U.S. Food and Drug Administration (2026a). 21 CFR Part 58.195: Good laboratory practice for nonclinical laboratory studies — Record retention. Electronic Code of Federal Regulations. URL: https://www.ecfr.gov/current/title-21/chapter-I/subchapter-A/part-58/subpart-K/section-58.195.
U.S. Food and Drug Administration (2026b). 21 CFR Part 11: Electronic records; electronic signatures. Electronic Code of Federal Regulations. URL: https://www.ecfr.gov/current/title-21/chapter-I/subchapter-A/part-11.
up to data specializes in regulatory study data management for pharmaceutical and life sciences companies. For over 20 years, the company has supported bioanalytical laboratories with automated solutions that eliminate data silos, ensure full regulatory compliance, and reduce manual overhead across the data lifecycle. The StudyPlus Archive Module is the company’s purpose-built solution for long-term retention of Watson™ studies. It is vendor-qualified under GAMP 5, validated against Watson™ versions 7.3, 7.4, and 7.6, and developed by an ISO 9001:2015 certified organization.
This content might also be engaging for you!
-
Expert Coffee Break Long-Term Study Archiving Recording
Blog Long-term Study Archiving After Watson LIMS™: Key Insights for Regulated Bioanalytical Labs In a regulated bioanalytical lab, the…
-
The Post-Merger Watson™ Problem: How to Phase Out Watson LIMS™ Cost-Effectively
Blog The Post-Merger Watson LIMS™ Problem: A Cost-Effective Path to Decommissioning A CFO/CIO guide to decommissioning legacy bioanalytical LIMS…
-
Four Dimensions of Time Savings in GxP Bioanalytical Study Reporting
Blog Four Dimensions of Time Savings in GxP Bioanalytical Study Reporting With rising expectations under ICH M10, FDA Part 11, and EMA…