Blog
Four Dimensions of Time Savings in GxP Bioanalytical Study Reporting
With rising expectations under ICH M10, FDA Part 11, and EMA guidelines, bioanalytical labs face growing pressure: more data, more documentation, and less room for error. Manual reporting still risks transcription mistakes, slows QC cycles, and complicates inspection readiness.
This article highlights four dimensions where automation drives measurable impact — data handling, validation loops, document assembly, and compliance readiness — helping labs reduce manual effort, ensure data integrity, and deliver submission-ready reports faster.
Executive Introduction: The Evolving GxP Landscape
In modern drug development, bioanalytical study reports are more than just technical deliverables. They are also crucial regulatory milestones that support decisions regarding safety, efficacy, and bioequivalence. The International Council for Harmonization (ICH) M10 guideline, finalized in 2022, has standardized expectations for bioanalytical method validation and study sample analysis across regions. This guideline provides detailed guidance on documentation and reporting.
The FDA’s recent draft guidance on electronic systems, electronic records, electronic signatures, and data integrity for in vivo bioavailability and bioequivalence (BA/BE) studies underscores regulators’ growing emphasis on data integrity, traceability, and reconstructability throughout the data lifecycle. Data that cannot be shown to be accurate, complete, and attributable may be considered unreliable for regulatory decision-making purposes.
Therefore, treating manual reporting errors as “mere inefficiencies” is unacceptable. Each manual transfer, unlogged correction, or undocumented run decision causes delays and regulatory risks. This article identifies four areas where manual “time sinks” in GxP bioanalytical reporting conflict with modern expectations and explains how structured automation can help ensure compliance and faster timelines.
Dimension 1 – Data Capture and Aggregation
In bioanalytical labs, a significant amount of time is spent on reporting work before any tables are written. This time is spent gathering data from instruments, LIMS, and spreadsheets into a unified, analyzable dataset.
Manual Status Quo
In many small and mid‑sized laboratories, the current manual process involves exporting results from chromatographic systems or ligand-binding platforms into spreadsheets. These results are then enriched with sample information from external files and manually converted into validation or study summary tables. Each cut-and-paste step creates potential “breaks” in data lineage and makes it more difficult to prove that the results are linked to specific samples, methods, operators, and instruments.
Regulatory Expectations for Electronic Source Data
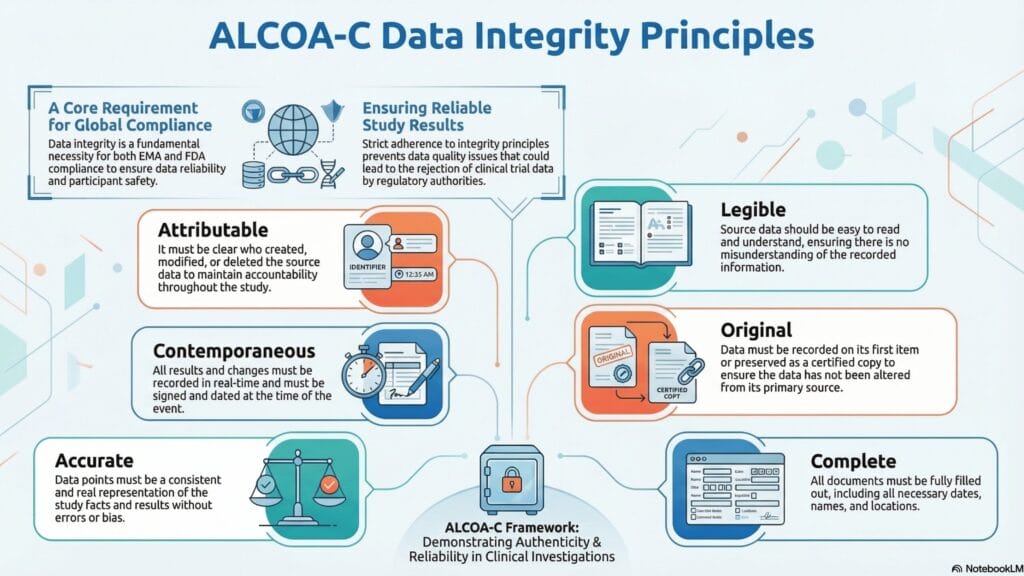
Recent FDA and EMA communications emphasize that electronic data used in clinical investigations and regulatory submissions must be collected and managed to maintain key attributes of data integrity—often summarized as ALCOA or ALCOA+. In the FDA’s draft guidance on electronic systems, records, and signatures, the agency emphasizes the need for:
- Validated electronic systems that reliably capture and retain data.
- Time-stamped audit trails that record creation and modification.
- The ability to generate accurate and complete copies of electronic records in a human-readable form.
Similarly, the FDA’s 2024 guidance document on data integrity for in vivo bioavailability/bioequivalence (BA/BE) studies reaffirms that source data should be complete, recorded at the time of the event, traceable to the responsible individual, and that sponsors understand and monitor how data transfer occurs between systems.
Infographic 1: ALCOA-C Data Integrity Principles
Time‑saving Potentials of Automated Data Capture
When bioanalytical data flows directly from instruments to a sample management system, several manual activities and associated verification loops can be reduced or eliminated.
- Fewer manual transfers: Cutting down on copy-and-paste steps reduces the need for thorough secondary checks on transcription and formatting.
- Built-in lineage: Automated import pipelines can attach metadata (sample IDs, run IDs, methods, operators) at the point of capture, supporting the “Attributable” and “Original” aspects of ALCOA+ without manual reconstruction.
- Near real-time aggregation: Electronic pathways enable near real-time consolidation of multiple runs and batches, reducing the time from analysis completion to the availability of reviewable datasets.
In this article, this aspect can be shown with a before-and-after comparison: hours spent manually reconciling spreadsheets versus minutes spent on automated ingestion and aggregation.
Dimension 2 – Validation and QC Review Loops
The FDA guideline details how to design, analyze, and document calibration curves, quality control samples, and stability experiments, which increases complexity under ICH M10. It provides:
- Acceptance ranges for accuracy and precision (e.g., typically ±15% for most levels, with broader limits at the lower limit of quantification [LLOQ]).
- Requirements to assess within-run and between-run performance across multiple runs.
- Expectations involving failed runs, repeat analyses, and reintegrations are captured, assessed, and documented.
Industry analyses, such as recent white papers on the implementation of ICH M10, emphasize that these expectations can greatly increase the volume of data to be evaluated and the number of scenarios in which run-level decisions must be justified.
Manual Pain Points
In manual workflows, analysts typically:
- Export calibration and QC data into spreadsheets.
- Calculate means, standard deviations, and coefficients of variation (%CV) for each level.
- Compare the observed accuracy and precision to the established acceptance criteria.
- Mark injections or runs as accepted or rejected, and update summaries when new runs are added.
Each extra run or correction can trigger a chain of updates across related spreadsheets and tables, lengthening cycle times and increasing the likelihood of calculation or linkage errors.
Automation as a Consistency and Time‑saving Engine
A data model that encodes validation rules and run acceptance logic can:
- Apply sponsor‑defined acceptance criteria consistently and immediately after each run is processed.
- Flag out‑of‑limit values and runs in real time, reducing the back‑and‑forth between analysts and reviewers.
- Recalculate summary statistics across all runs automatically when additional data are added, eliminating manual rework.
Laboratories can shorten validation cycles and focus scientific review efforts on borderline or anomalous cases rather than routine calculations by reducing manual recalculation and reassembly.
Dimension 3 – Document Authoring and Report Assembly
Producing submission-ready bioanalytical reports requires more than just technical accuracy. It also involves adhering to specific structural and content requirements.
Structured Reporting under ICH M10
ICH M10 describes the expected content of both method‑validation and study sample analysis reports, including:
- Descriptions of methods, matrices, calibration ranges, and key validation parameters.
- Tables summarising accuracy, precision, and stability at each QC level, within and between runs.
- Listings of calibration curve performance.
- Documentation of study sample analyses, including failed runs, repeat analyses, and deviations.
Recent implementation comments indicate that the practical impact of M10 on some comparative bioavailability/bioequivalence (BA/BE) studies is a significant increase in the amount and detail of information that must be documented, especially for failed or reprocessed runs that might previously have been summarized more concisely.
Manual Assembly Challenges
In a manual environment, assembling these reports can require:
- Compiling data from multiple spreadsheets and instruments into Word tables.
- Maintaining consistent formatting and formulas across dozens of tables and appendices.
- Ensuring that every run mentioned in narrative sections is correctly reflected in summary and listing tables.
- Updating multiple document sections when late corrections or additional runs are introduced.
It is common for teams to spend days or weeks finalizing a single comprehensive report, with multiple cycles of formatting, cross‑checking, and version consolidation.
Template‑driven, Automated Authoring
Automated reporting frameworks can significantly compress this timeline by:
- Generating validation and study tables directly from a central data store according to pre‑defined templates aligned with ICH M10’s structures and internal SOPs.
- Populating narrative sections with key parameters (e.g., number of runs, acceptance rates) from the database to reduce manual duplication.
- Regenerating complete reports or sections automatically when the underlying data changes without the need for manual copy-and-paste.
This reduces document preparation time and lowers the risk of internal inconsistencies between data listings, summary tables, and narrative descriptions. Our article, “Beyond Manual Reporting: A Sneak Peek into the Future of Automated Study Data Reporting,” shows how Word-based report templates can automatically reduce document creation time by up to 70% while generating GxP-compliant reports suitable for regulatory submission. This aligns with ICH M10’s focus on structured, comprehensive reporting, freeing up analysts to focus on more important tasks.
Dimension 4 – Inspection Readiness and Data Integrity
The value of efficient reporting is ultimately tested during regulatory inspections and sponsor audits. The ability to trace an analytical result’s history, from sample receipt to final reported concentration, is crucial to data integrity.
Inspection Focuses on Reconstructability
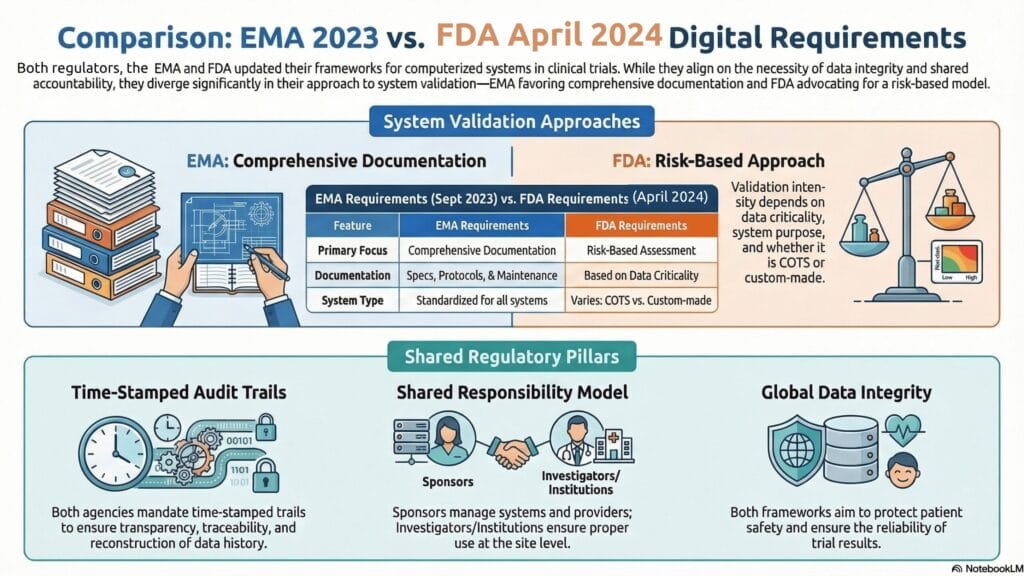
The FDA’s recent draft guidance on electronic systems, records, and signatures emphasizes that agencies will review not only final datasets but also supporting documentation, such as system validation records, audit trails, and user access management records. The 2024 data integrity guidance for in vivo bioavailability and bioequivalence studies further emphasizes that sponsors should be able to understand and monitor how data were collected, processed, and transferred between systems.
EMA’s recent discussions on computerized systems and electronic data suggest a consistent approach involving validated systems, appropriate user controls, and audit trails that enable reconstruction of event sequences.
Limitations of Manual or Semi‑electronic Systems
When key decisions and changes are recorded only in emails, paper notebooks, or ad hoc spreadsheet comments, reconstructing a complete data history can be time-consuming and error-prone. Gaps or inconsistencies in this reconstruction may be viewed as weaknesses in the quality management system, even if the underlying science is sound.
Automation as a Driver of Inspection Readiness
An integrated, automated reporting environment can support inspection readiness by:
- Maintaining timestamped audit trails for essential actions, such as data entry, modification, and approval, linked to individual user accounts.
- Providing consistent, query-ready views of how runs were evaluated, which ones were rejected, and why.
- Enabling quick updates to reports and listings showing the current, approved dataset while retaining historical versions.
From a time-saving perspective, this results in less ad hoc reconstruction before and during inspections, fewer manual reconciliations, and a more predictable inspection preparation workload.
As detailed in the sneak peek article, StudyReporter’s end-to-end data process—from raw bioanalytical data to live tables and submission-ready reports—was designed to maintain traceability at every stage. Every reported value can be traced back to its source data, which supports the reconstructability of results that regulators expect during inspections and sponsor audits. Rather than assembling this traceability after the fact from spreadsheets and emails, the system maintains it continuously as part of routine reporting.
Infographic 2: Comparing EMA and FDA Electronic System Requirements
Conclusion: Automation as an Enabler of Compliance and Time Savings
Recent FDA and EMA guidance, as well as ICH M10, indicate that the future of bioanalytical study reports will require greater structure and transparency than ever before. In this environment, relying primarily on manual processes creates a double burden: increased data integrity risks and longer reporting timelines.
Beyond the conceptual arguments presented in this article, practical implementations demonstrate what this shift can look like in everyday operations. As outlined in “Beyond Manual Reporting: A Sneak Peek into the Future of Automated Study Data Reporting,” automated filling of structured Word templates and end-to-end data workflows can significantly reduce the effort required for document creation while maintaining complete traceability from source data to submission-ready reports. This real-world example shows how the four dimensions discussed here provide measurable benefits to laboratories of various sizes.
Automated, GxP-aligned data platforms that address the four key dimensions “data capture and aggregation”, “validation and QC review loops”, “document authoring and assembly”, and “inspection readiness and data integrity“ provide a practical way forward. While they cannot replace scientific judgment or the need for clear SOPs, they can reduce manual transcription and calculation, apply acceptance rules and reporting structures consistently, and provide the audit trails and reconstructability that modern inspections require.
Infographic 3: Mastering GxP Compliance with StudyReporter

References
Clinical Pathways Research (2024). FDA Releases New Draft Guidance on Data Integrity for In Vivo Bioavailability and Bioequivalence Studies. URL: https://www.clinicalpathwaysresearch.com/blog/2024/7/5/fda-releases-new-draft-guidance-on-data-integrity-for-in-vivo-bioavailability-and-bioequivalence-studies.
Covington & Burling LLP (2023). FDA Releases Draft Guidance on Electronic Systems, Records, and Signatures in Clinical Investigations. URL: https://www.cov.com/en/news-and-insights/insights/2023/04/fda-releases-draft-guidance-on-electronic-systems-records-and-signatures-in-clinical-investigations.
EMA (2023). Guideline on the Packaging Information of Medical Products for Human Use Auhrorised by the Union. September 2023. URL: https://health.ec.europa.eu/system/files/2023-09/2018_packaging_guidelines_en_1.pdf.
EMA (2022). ICH M10 on bioanalytical method validation – Scientific guideline. URL: https://www.ema.europa.eu/en/ich-m10-bioanalytical-method-validation-scientific-guideline.
FDA (2024). Guidance Document – Data Integrity for In Vivo Bioavailability and Bioequivalence Studies. URL: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/data-integrity-in-vivo-bioavailability-and-bioequivalence-studies.
Marciulioniene, V., & Hulbert, J. (2023). Confidently Compliant? Considerations for Industry Players to Meet the New EMA Guideline. In Clinical Edge, Issue 1, S. 12-13. URL: https://clinicaledge.xtalks.com/magazine/issue1/emas-new-framework.
Saunders, S. (2023). FDA Part 11 New Draft (March 2023) Guidance. Florence Healthcare. URL: https://www.florencehc.com/blog-post/fda-part-11-new-draft-march-2023-guidance.
up to data has been supporting pharmaceutical and life sciences companies with automated laboratory processes for regulatory study data management for over 20 years. Our solutions eliminate data silos, implement secure automated data transfer processes, and reduce manual activities while ensuring full regulatory compliance.
PDF Article Download
This content might also be engaging for you!
-
The Post-Merger Watson™ Problem: How to Phase Out Watson LIMS™ Cost-Effectively
Blog The Post-Merger Watson LIMS™ Problem: A Cost-Effective Path to Decommissioning A CFO/CIO guide to decommissioning legacy bioanalytical LIMS…
-
Long-Term Retention of Bioanalytival Watson LIMS studies: A Compliance-First Perspective
Blog Long-Term Retention of Bioanalytical Watson LIMS™ Studies: A Compliance-First Perspective Most bioanalytical labs running Watson LIMS™ have never made a…
-
Beyond Manual Reporting | Sneak Peak StudyReporter
Blog Beyond Manual Reporting: A Sneak Peek into the Future of Automated Study Data Reporting Discover in our exclusive…