Blog
From Raw Data to Decision: Why Automated Reporting is Essential for GxP Labs
Bioanalytical reporting involves converting raw analytical data from validated methods into structured, submission-ready documentation for regulatory purposes or sponsor decision-making. This process links each sample, chromatogram, or assay signal to the final submitted concentration or pharmacokinetic (PK) parameter, enabling traceability, and auditability for regulators.

What Do Global Guidelines Actually Require?
Global guidelines, such as ICH M10 and the FDA Bioanalytical Method Validation (BMV) Guidance, explicitly cover not only method validation and study sample analysis but also documentation and reporting requirements. A complete bioanalytical report typically comprises:
- A method section describing the matrix, calibration range, sample preparation, analytical conditions, and key validation parameters (selectivity, accuracy, precision, and stability).
- Validation results consolidated into tables showing calibration curve performance, within- and between-run accuracy and precision at all QC levels, and stability data.
- Study sample analysis documentation, including analytical run listings, sample concentration results, and records of repeat analyses, reinjections, and deviations from the predefined plan.
- Summaries suitable for the Common Technical Document (CTD).
Without this documentation, even technically sound assays lack the traceability and transparency that regulators require when making decisions about safety, efficacy or bioequivalence.
Why Do Sponsors and Regulators Insist on Comprehensive Reporting?
From a regulatory and sponsor perspective, bioanalytical reports address a key question:
“Can these concentrations be trusted as the basis for clinical or regulatory decisions?”
ICH M10 states that all method validation data and analytical results supporting regulatory submissions must be fully documented, including failed runs and out-of-specification quality control (QC) tests. The FDA guidance devotes a separate section to documentation and reporting, providing example tables, and matrices to illustrate the required information.
From their perspective, a compliant bioanalytical report must include the following:
- Traceability: A clear chain from sample receipt, storage, and preparation through analysis to the reported result, including any reanalysis decisions and their justification.
- Data integrity: Complete, consistent, and accurate data with an audit trail that makes changes and duplicate work visible.
- Reviewability: A structure that enables reviewers to quickly understand the method, assess how the acceptance criteria were met, and evaluate the reliability of the data.
They even go one step further by emphasizing the need for harmonized report structures and tables of contents for both validation and study reports. However, this harmonization only works if reporting is systematic, standardized and complete at the data-generation level.
Life in Excel Spreadsheets: Manual Reporting in Small Labs and CROs
Despite these clear expectations, many small and mid-sized bioanalytical laboratories and contract research organizations (CROs) lack a comprehensive Laboratory Information Management System (LIMS). Instead, they rely on a variety of systems:
- Excel spreadsheets for sample lists, randomization codes and tracking.
- Instrument-specific software (e.g. LC-MS/MS or ligand-binding platforms) for quantitative analysis and peak integration.
- Additional spreadsheets or point tools for PK calculations and summary tables.
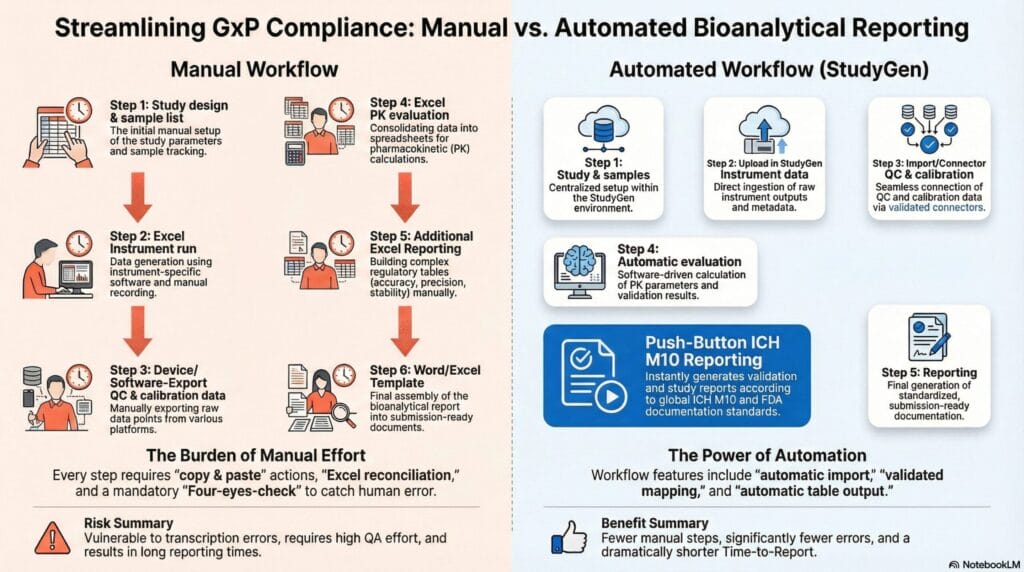
In this environment, producing a GxP-ready bioanalytical report requires manual data consolidation. Sample IDs must be reconciled across multiple Excel files and instrument exports. Calibration and QC results must be copied into validation summary tables that mirror the examples provided by ICH M10 and the FDA BMV (e.g., back-calculated standards, QC accuracy and precision, stability summaries). Study sample analysis listings are compiled run by run, including failed runs, re-runs, and reinjections, in a style consistent with the FDA appendices.
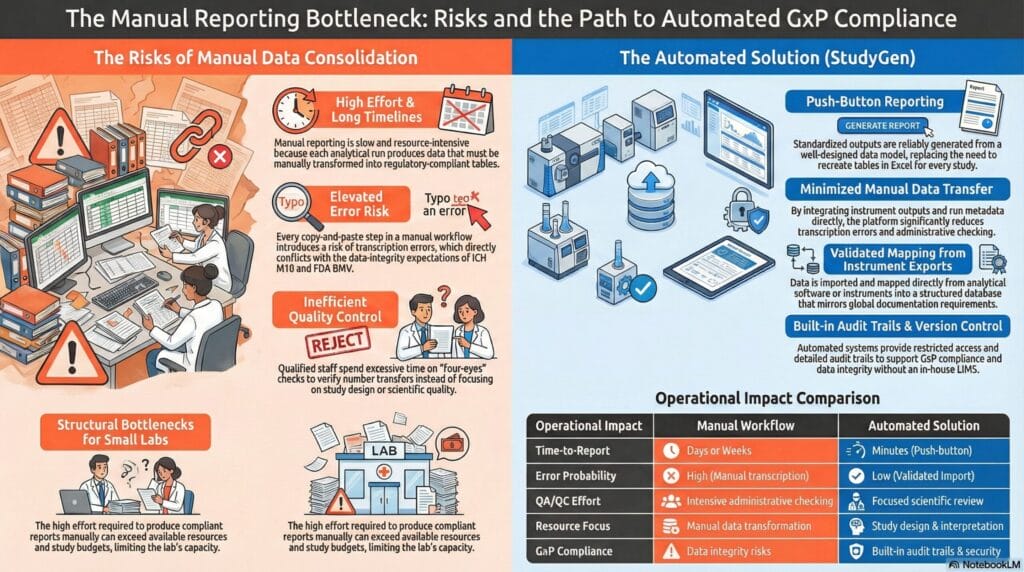
This manual workflow has predictable drawbacks:
- High effort and long timelines – It is slow and resource-intensive because each accepted run produces data that must be manually transformed into regulatory tables.
- Elevated error risk – Every copy-and-paste step introduces a risk of transcription errors, which conflict with the data-integrity expectations under ICH M10 and FDA BMV.
- Inefficient quality control – The four-eyes principle is time-consuming, as highly qualified staff spend hours verifying that numbers have been transferred correctly rather than focusing on scientific quality or study design.
For smaller organizations, this can create a structural bottleneck: although technically robust data are available, the effort required to produce compliant bioanalytical reports exceeds study budgets and available resources.
What Regulators Expect to See – and Why This Matters for Automation
A look at ICH M10 and FDA BMV shows how structured bioanalytical reporting has evolved.
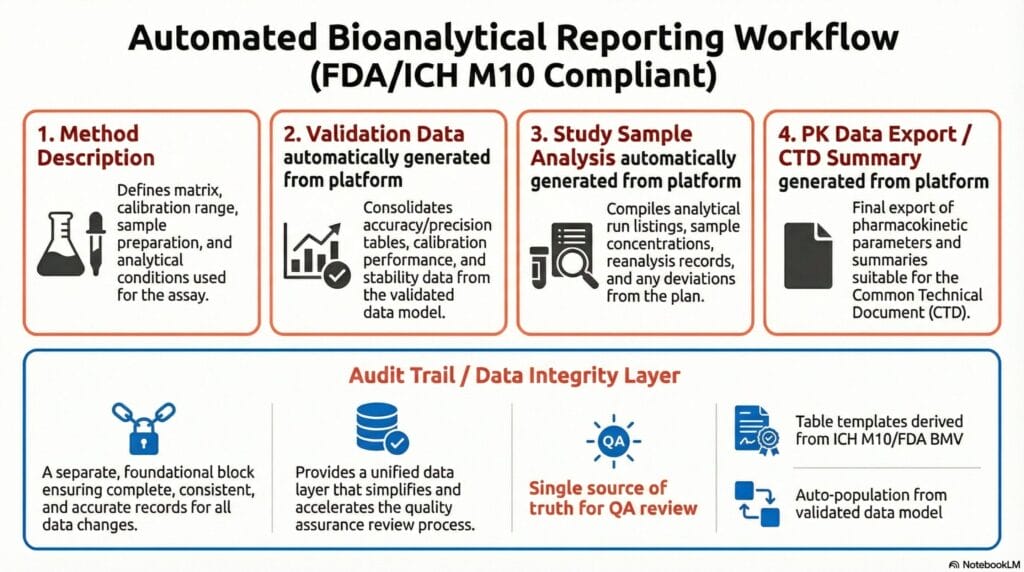
Common table types include:
- Validation summary tables: accuracy, precision and stability at each QC level, both within and between runs.
- Calibration curve performance: nominal vs. back-calculated concentrations for all standards in each accepted run.
- Analytical run listings: per-run overviews of all calibrators, QCs and samples, including failed runs and re-runs.
- Sample concentration listings: subject/time-point concentrations exported for PK analysis.
ICH M10 includes a dedicated section on documentation and recommended content for validation and study sample reports, with example tabular formats. Similarly, the FDA guidance provides tables and checklists in its appendices that specify which information should be recorded and retained.
This high degree of structure is exactly why automation is such a powerful tool. If the expected outputs are standardized, they can be reliably generated from a well-designed data model rather than being manually recreated in Excel for each study.
Turning Reporting into a Push-Button Process with Cloud Data Platforms
For laboratories and CROs without a LIMS, a cloud-based bioanalytical data platform offers a pragmatic alternative to on-premises IT infrastructure. Rather than replacing every existing system, it integrates them:
- Instrument outputs and run metadata are imported from existing analytical software or the instrument itself.
- Sample information and study design details can be imported from Excel spreadsheets or other local tools.
- Validation and in-study runs are stored in a central, structured database that mirrors the documentation requirements of ICH M10 and FDA BMV.
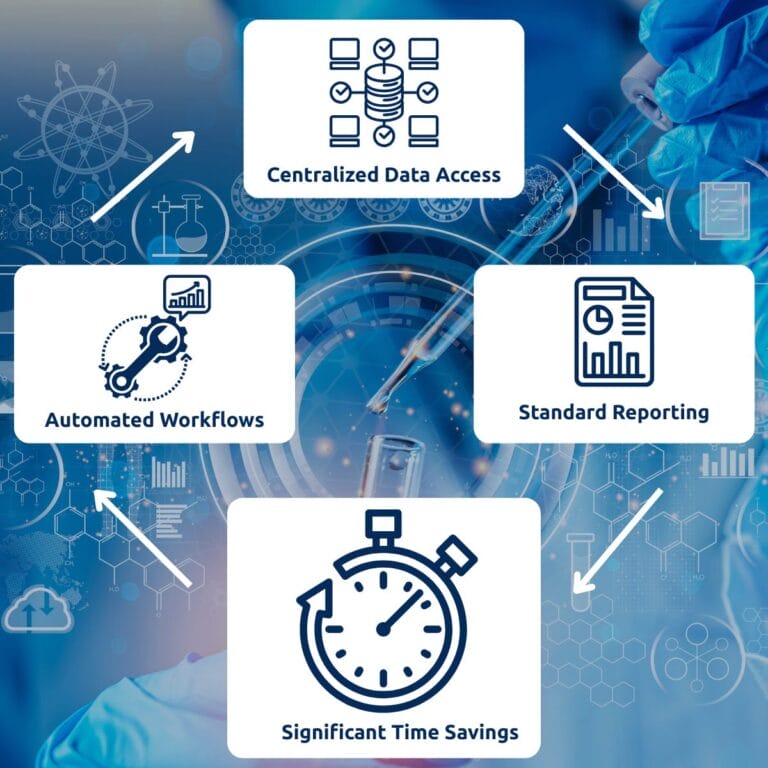
On top of this unified data layer, an automated reporting process can be defined to:
- Generates validation summary tables, calibration curve performance tables, QC acceptance overviews, and study sample listings in line with the structures illustrated in ICH M10 and FDA BMV.
- Minimizes manual data transfer, thereby significantly reducing transcription errors and the need for purely administrative four-eyes checks.
- Provides audit trails, version control, and restricted access to support data integrity and GxP compliance, thereby eliminating the need for a full in-house LIMS implementation.
Rather than spending days or weeks consolidating spreadsheets, teams can focus on study design, method optimiuzation and scientific interpretation. Reporting timelines shorten, and consistency and inspection readiness improve.
Conclusion: Where StudyGen 360 Comes into Play
StudyGen 360 is designed for this scenario: labs and CROs with heterogeneous tools but clear GxP reporting obligations. Rather than forcing a complete system replacement, StudyGen 360 serves as a cloud-based data and reporting layer on top of your existing environment.
In practice, this enables you to:
- Consolidate all relevant study and analytical data on a single, secure cloud platform, eliminating the need to build and qualify complex local infrastructure.
- Implement automated, ICH M10-compliant bioanalytical reporting aligned with FDA documentation requirements.
- Expand your service offerings to include full GxP bioanalytical reporting while reducing manual effort, error risk and linear headcount growth.
For smaller GxP labs and CROs, automated bioanalytical reporting is not merely a technical upgrade. It is also a strategic enabler, transforming existing data into inspection-ready, regulatory-compliant documentation without requiring significant IT infrastructure.
References
European Medicines Agency (EMA), 2022, “ICH guideline M10 on bioanalytical method validation and study sample analysis”, EMA, Amsterdam. Available at: https://www.ema.europa.eu/en/documents/scientific-guideline/ich-guideline-m10-bioanalytical-method-validation-step-5_en.pdf.
US Food and Drug Administration (FDA), 2018, “Bioanalytical Method Validation – Guidance for Industry”, FDA, Silver Spring, MD. Guidance page: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/bioanalytical-method-validation-guidance-industry (Accessed 4 February 2026). PDF: https://www.fda.gov/files/drugs/published/Bioanalytical-Method-Validation-Guidance-for-Industry.pdf.
Verhaeghe, T, Dudal, S, Mahler, HC, et al., 2014, “Recommendations from the Global Bioanalysis Consortium Team A8: Documentation”, The AAPS Journal, vol. 6, no. 4, pp. 437–446. Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3933576/.
up to data has been supporting pharmaceutical and life sciences companies with automated laboratory processes for regulatory study data management for over 20 years. Our solutions eliminate data silos, implement secure automated data transfer processes, and reduce manual activities while ensuring full regulatory compliance.
This content might also be engaging for you!
-
Beyond Manual Reporting | Sneak Peak StudyReporter
Blog Beyond Manual Reporting: A Sneak Peek into the Future of Automated Study Data Reporting Discover in our exclusive…
-
CRO-Sponsor Collaboration: Digital Risk Mitigation
Blog Optimizing CRO-Sponsor Collaboration: Mitigating Risks in Bioanalytical Study Reporting Manual processes in bioanalytical studies jeopardise efficiency, data integrity,…
-
Context-First AI Approach for CROs | Multi-Run Assay Control
Blog The Critical Role of Multi-Run Assay Control in Bioanalytical Research: A Context-First AI Approach for Contract Research Organizations…